
上海百赛生物创建于2005年,专注于生命科学行业,为客户提供耗材、试剂、仪器、技术等相关服务及解决方案,不仅拥有多个国际知名品牌在中国的独家代理或者一级代理,还不断拓展自主品牌产品。

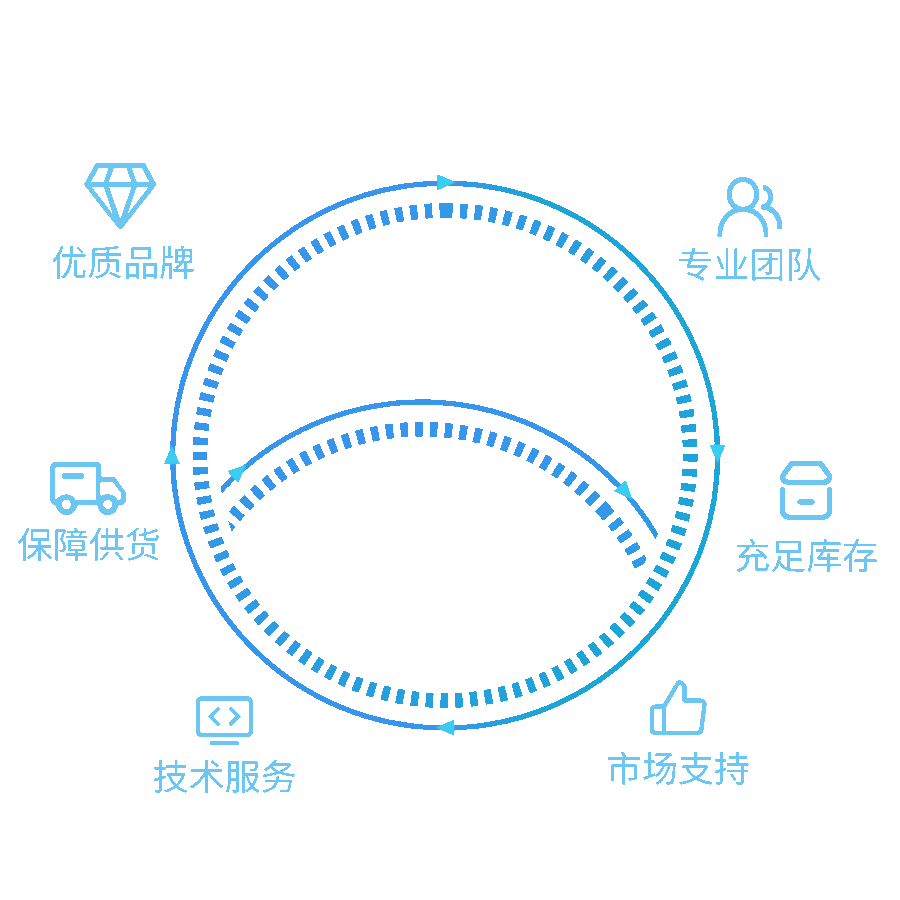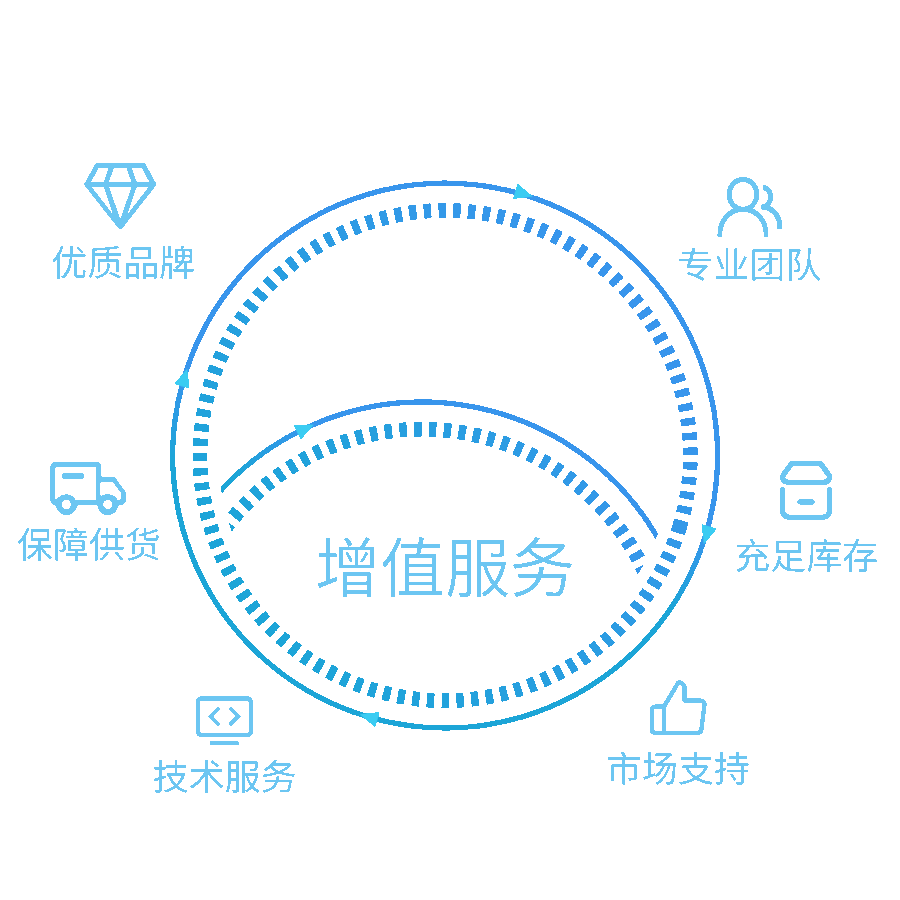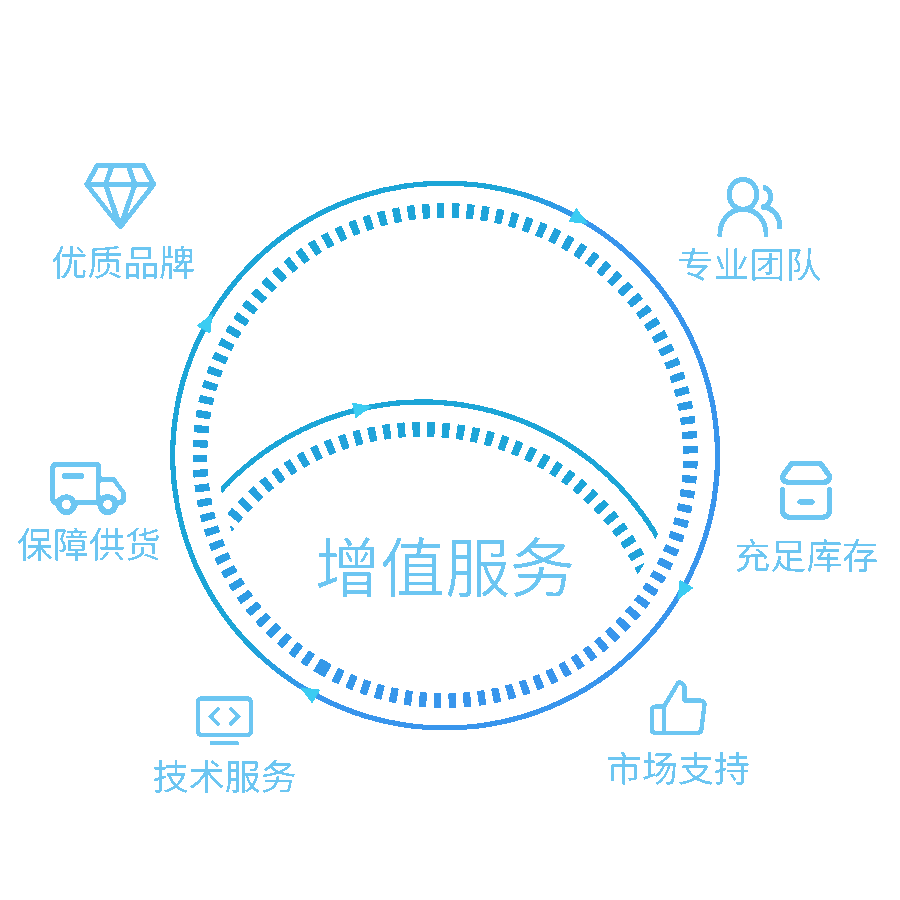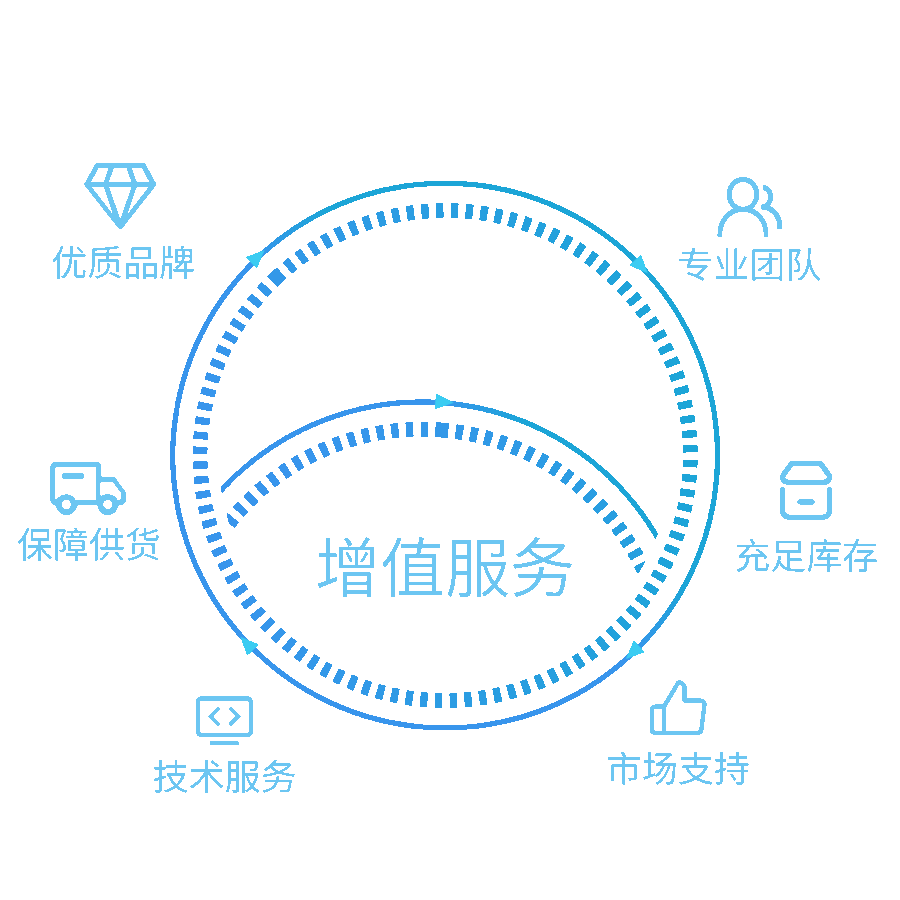
我们的解决方案
提供实验室一站式服务






05
2005年
注册成立
07
2007年
签约AXYGEN中国区代理

10
2010年
在北京、广州、深圳、武汉、重庆、成都、西安分别建立分公司,成为一家真正意义上的全国性公司

12
2012年
确立集耗材、试剂、仪器三条产品线的经营模式,致力于为客户提供实验室一站式服务

16
2016年
建立华东、华北、华南、三大物流仓储基地

18
2018年
与上海皓嘉合并,开始代理Takara试剂

20
2020年
成为丹纳赫旗下多品牌在中国区域代理及战略合作伙伴
成功完成首轮融资,开始资本化发展
成功完成首轮融资,开始资本化发展
谁选择了我们? —— 我们的合作伙伴
顾客购买的不仅仅是产品,更是服务态度和服务精神,感谢所有客户对百赛生物的认可和支持!